
Biologics such as monoclonal antibodies and vaccines succeed or fail based on precise molecular recognition. For this reason, epitope mapping – pinpointing the exact binding sites on target antigens – has become a strategic tool in biologics R&D.
In this article, we explain the two major classes of epitopes (linear vs. conformational) and how each can be identified using state-of-the-art techniques. We also compare epitope mapping methods in terms of resolution and complexity, and discuss strategic implications for antibody engineering, vaccine design, biosimilar development, and immunogenicity risk assessment.
What’s the difference between linear and conformational epitope mapping?
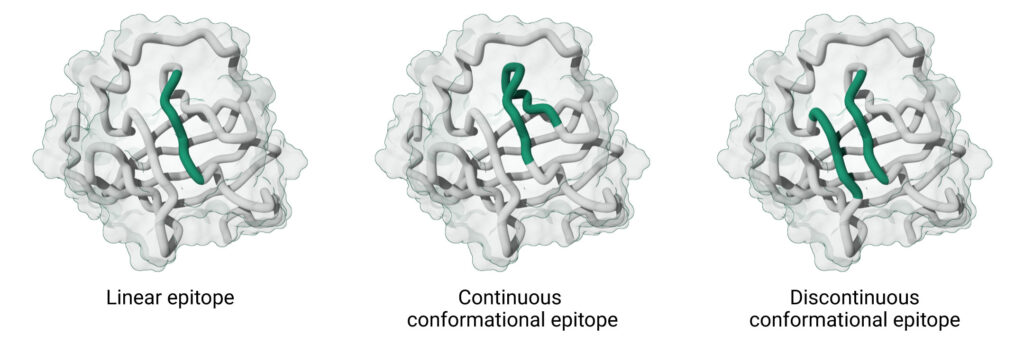
An epitope is the specific region of an antigen recognized by an antibody’s binding site. Epitopes are commonly categorized as either linear or conformational, but the reality is a bit more nuanced.
Linear epitopes are continuous peptide segments in the primary sequence of the antigen, typically short stretches of 5–20 amino acids that remain antigenic even in a denatured state.
In contrast, conformational epitopes are recognized based on their three-dimensional shape on the folded antigen. This class includes two subtypes:
- Continuous conformational epitopes, which are composed of a single stretch of amino acids whose structure is required for antibody recognition. Though the sequence is continuous, the epitope is recognized by its antibody when it adopts a specific 3D conformation.
- Discontinuous conformational epitopes, which are formed by amino acids that are distant in the primary sequence but are brought together through protein folding. These are typically found on well-folded, globular proteins.
In practical terms, linear epitopes are often found in flexible or exposed regions such as loops or termini (and even on polysaccharides or nucleic acids). They can often be identified by antibodies even if the antigen is unfolded (e.g. in a Western blot under denaturing conditions). Conformational epitopes, on the other hand, are usually found on the surfaces of well-folded, globular proteins in their native conformation.
Although it’s often claimed that the vast majority of B-cell epitopes is conformational – with widely cited estimates suggesting that ~90% are conformational – this figure originates from a small, early dataset of antibody–antigen crystal structures published in 1986. That dataset was heavily biased towards high-affinity monoclonal antibodies, antigen type and assay format, and doesn’t represent the diversity of antibody responses seen in infectious diseases, autoimmunity, anti-drug responses, or vaccine research. In reality, the proportion of linear versus conformational epitopes varies greatly depending on the antigen, the immune context, and the experimental methods used. Despite this, the claim that ‘only 10% of epitopes are linear’ has been widely repeated, perpetuating the misconception that linear epitope mapping is not useful. In reality, linear epitope mapping remains a powerful and highly informative approach, particularly when integrated with complementary techniques. It offers unique advantages in scalability, reproducibility, and ease of interpretation, making it an indispensable tool in antibody characterization, vaccine development, and diagnostic discovery.
Linear and conformational epitopes each demand different mapping approaches. Linear epitopes can often be mapped by simple peptide-based experiments, while conformational epitopes may require higher-order structural analysis. From a strategic standpoint, knowing whether a critical epitope is linear or conformational guides the choice of mapping techniques and informs downstream decisions (e.g. whether a vaccine antigen must maintain a specific folded structure or if an antibody can be screened by peptide assays). In the next sections, we discuss the toolbox of experimental methods for mapping each type of epitope.
The different mapping challenges for linear and conformational epitopes
Linear epitopes can often be identified by testing overlapping peptide fragments of the antigen sequence for antibody binding. Because linear epitopes remain recognizable even in isolated peptide form, mapping them is relatively straightforward and high-throughput using synthetic peptides.
Conformational epitopes, on the other hand, are either made up of continuous amino acid residues that form a specific structure, or are brought into proximity by the three-dimensional folding of the protein in the case of discontinuous epitopes. Traditionally, this has meant that conformational epitope mapping required more complex methods – such as crystal structure analysis, hydrogen-deuterium exchange mass spectrometry, mutagenesis, or cryo-electron microscopy – that probe the intact antigen in its native conformation.
However, recent advances have shown that conformational mapping does not always require such elaborate techniques. The use of cyclic constrained peptide libraries – similar to those displayed on linear peptide microarrays – can mimic the native structural motifs of a protein, allowing the identification of conformational epitopes with similar ease and throughput as linear mapping. For instance, studies using constrained peptide microarrays have successfully mapped the conformational epitope of rituximab, a therapeutic antibody, by finding which cyclic peptides were recognized by it. These peptides simply imitate the 3D shape of the epitope on the actual protein.
This constrained peptide array method offers a hybrid approach combining high-throughput workflows with the ability to capture conformational motifs, making conformational mapping more accessible and efficient.
High-throughput mapping of linear and conformational epitopes with peptide microarrays
Peptide microarrays are widely regarded as the method of choice for high-throughput antibody epitope mapping, especially for identifying linear epitopes. In a typical setup, overlapping linear peptides covering the antigen sequence are synthesized and immobilized on a solid surface, allowing thousands of peptide–antibody interactions to be screened in parallel. This approach has been successfully used to map epitopes at single-amino-acid resolution across entire proteomes, such as with the SARS-CoV-2 proteome microarray, which identified dozens of immunogenic regions recognized by patient antibodies.
While standard linear peptide arrays are excellent for detecting linear epitopes, many antibody targets depend on the native three-dimensional structure of the antigen. These conformational binding sites cannot be effectively captured using traditional linear peptides, as they require structural context provided by protein folding.
To overcome this limitation, PEPperPRINT has developed high-throughput peptide microarrays that incorporate libraries of cyclic constrained peptides. These peptides are structurally stabilized to mimic the shape and spatial conformation of native protein loops or motifs, enabling the detection of conformational epitopes using the same high-throughput platform traditionally used for linear mapping.
In the study on rituximab mentioned above, researchers conducted a conformational epitope mapping using PEPperPRINT’s microarrays containing both linear and constrained cyclic CD20 peptides. Interestingly, rituximab showed no binding to any of the linear peptides. However, strong and specific signals were observed for several constrained cyclic peptides, especially those containing the consensus motif EPANPSEK, confirming that the antibody recognizes a conformational epitope.
The mapping didn’t stop at detection. Using PEPperMAP® Epitope Substitution Scan, the team systematically substituted each amino acid in the epitope with all 20 standard amino acids to identify which positions were critical for binding. This allowed a high-resolution view of essential and conserved versus variable positions within the epitope.
Such detailed epitope profiles have wide-ranging implications. They enable assessment of potential antibody cross-reactivity and can identify sequence variations in patient populations that may affect therapeutic efficacy. For instance, detecting point mutations in conserved epitope regions could guide patient selection in clinical trials, reduce the immunogenicity of antibody drugs, or help explain differences in response rates, paving the way for more personalized antibody therapies. Furthermore, high-resolution epitope data can also support regulatory decisions, such as distinguishing a biosimilar from a reference monoclonal antibody.
In short, while peptide microarrays have long been associated with linear epitope mapping, the integration of cyclic constrained peptide libraries has transformed them into a versatile platform capable of capturing both linear and conformational epitopes, offering unmatched speed, resolution, and clinical insight.
Strategic considerations for epitope mapping technologies
When choosing epitope mapping techniques, it’s crucial to balance speed, throughput, and resolution to fit the project’s needs. Rapid screening methods like peptide microarrays provide quick answers about linear epitopes across the entire sequence of an antigen or even proteome, representing an excellent first pass for vaccine or antibody development where time is critical.
Mid-throughput biophysical methods (HDX-MS, proteolysis-MS, mutational scanning) offer the next layer of detail, mapping an antibody’s footprint on a native antigen without the delay of solving a crystal structure. These methods can often distinguish whether an antibody binds the epitope or overlaps with functional sites, which is strategically important for therapeutic antibody development.
On the other hand, if absolute atomic-level confirmation is required (for example, in regulatory filings or intellectual property claims), structural biology may be pursued in parallel or downstream, accepting the longer timeline for X-ray crystallography or cryo-EM to definitively visualize the epitope.
Given these considerations, peptide microarrays – particularly those incorporating both linear and constrained cyclic peptides, as developed by PEPperPRINT – offer a flexible, efficient entry point into epitope mapping workflows. They provide a high-throughput, high-resolution method capable of identifying both linear and conformational epitopes without the complexity or time investment of structural biology. This makes them well-suited not only for early-stage discovery, but also for downstream characterization efforts, such as epitope fine-mapping, immunogenicity profiling, or biosimilarity assessments.
In most cases, starting with peptide microarrays enables rapid generation of actionable data that can guide subsequent decisions – whether to prioritize a candidate, use the best research antibody, refine antibody engineering, or flag potential liabilities – before investing in slower, resource-intensive methods. For researchers and developers seeking speed, scalability, and structural insight within a single platform, this approach strikes an effective balance across scientific rigor and operational practicality.
Contact PEPperPRINT today to discuss how peptide microarrays can unlock deeper insights and drive your research to the next level.
← Go back